Dans ce chapitre, nous verrons des techniques qui nous permettent de déterminer si un jeu de données multivarié (qui contient plusieurs variables) contient des regroupements naturels de données.
On peut avoir différentes motivations pour chercher des regroupements dans les données. Les regroupements peuvent par exemple faciliter notre compréhension des données ou simplifier les processus de décisions. Ils agissent comme un préjugé : plutôt que de regarder tous les détails d’un échantillon, on peut le juger ou le comprendre uniquement par son appartenance à un groupe. Ce n’est pas très éthique d’agir ainsi lorsque l’on parle d’humains, mais le reste de la nature est moins soucieux de ce genre de problématique!
Nous verrons dans ce chapitre deux techniques de regroupements, soit l’analyse des K-means et la classification hiérarchique.
27.2 La technique des K-means
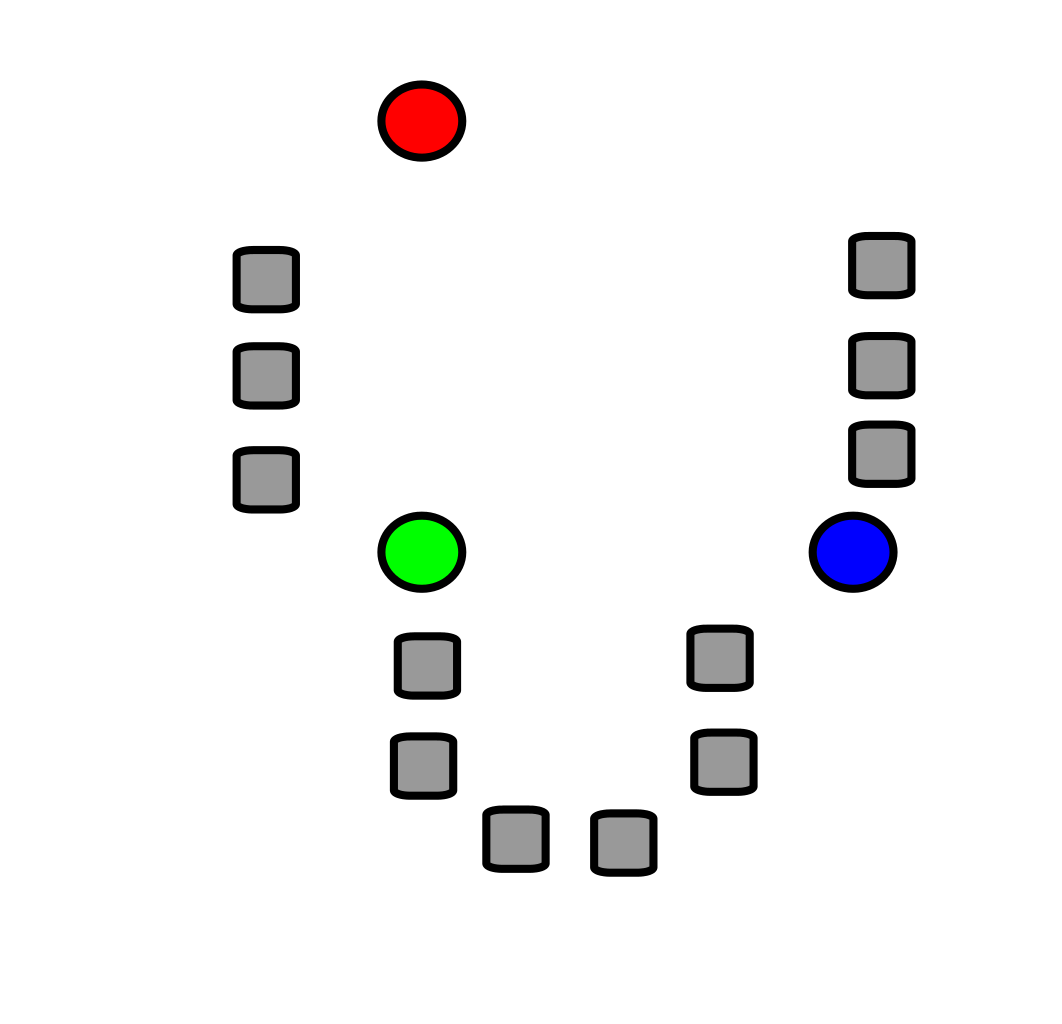
Pour présenter la technique des K-means, démarrons avec un scénario où nous avons observé une douzaine d’échantillons, sur lesquels nous avons mesuré deux variables, que nous présentons ici, une sur l’axe des X et l’autre sur l’axe des Y :
Si je vous demandais intuitivement combien de groupes contient ce jeu de données, vous répondrez probablement 3 groupes.
Mais où tracer précisément la ligne entre les groupes de façon objective?
27.2.1 Fonctionnement de l’algorithme
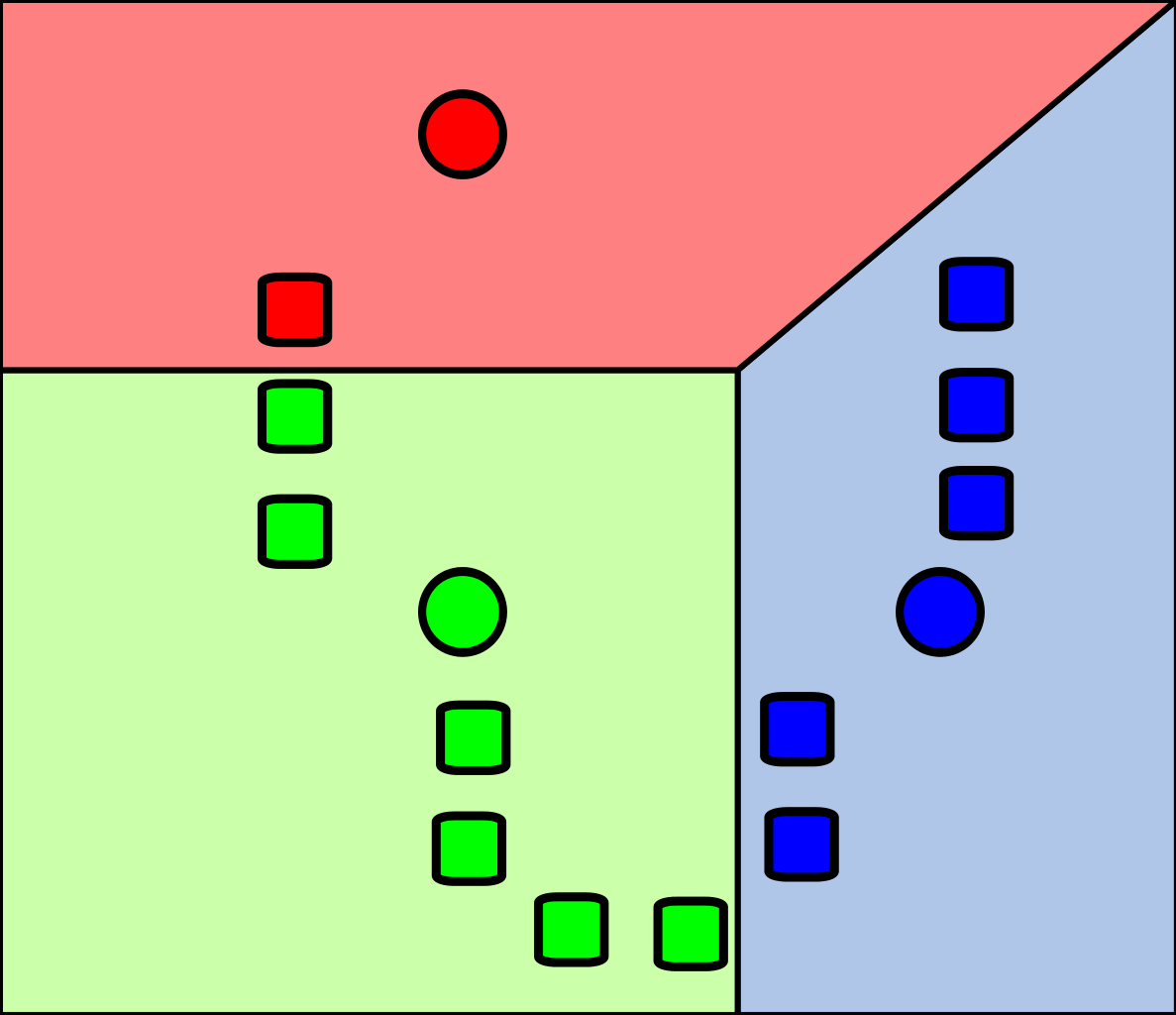
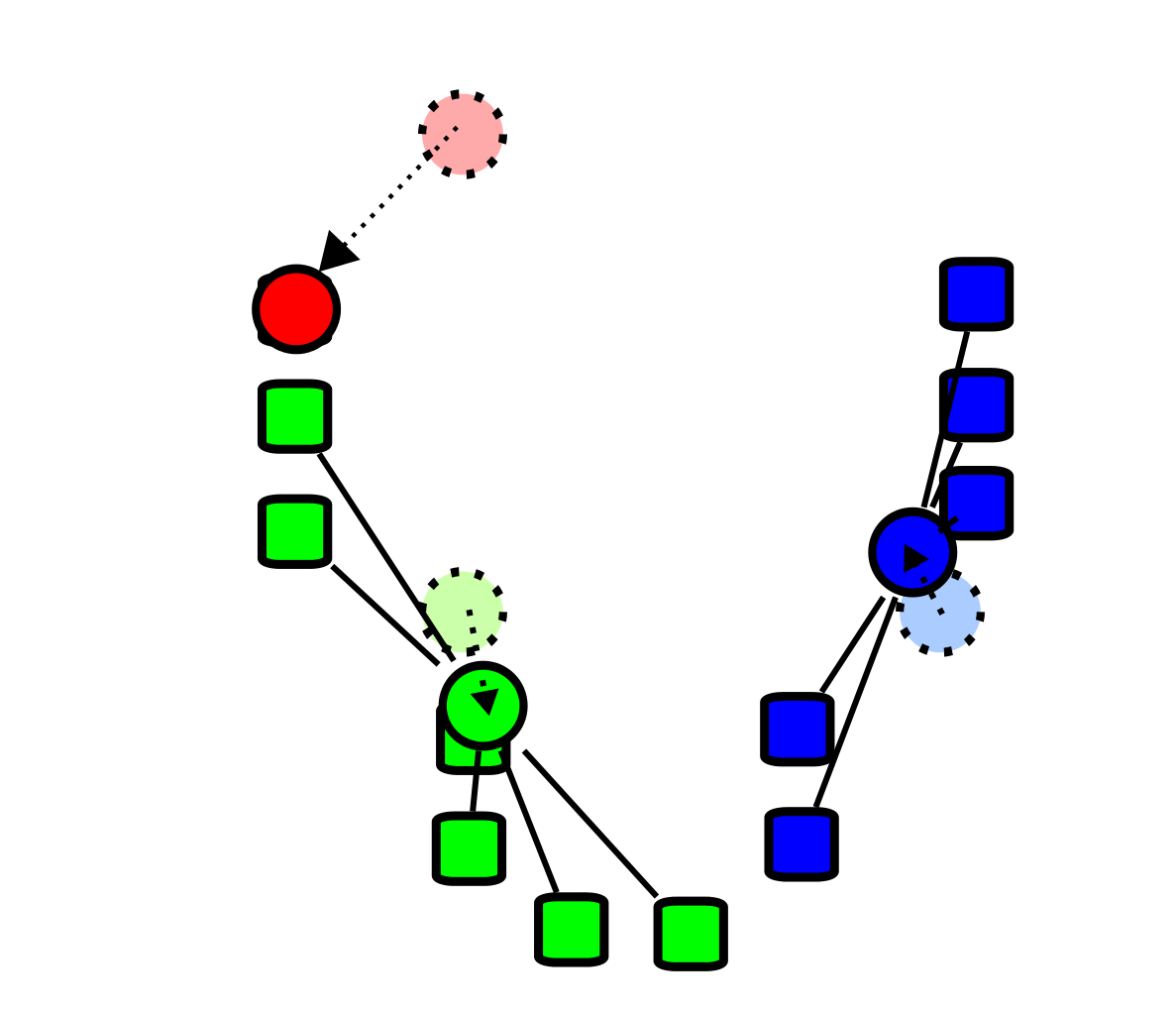
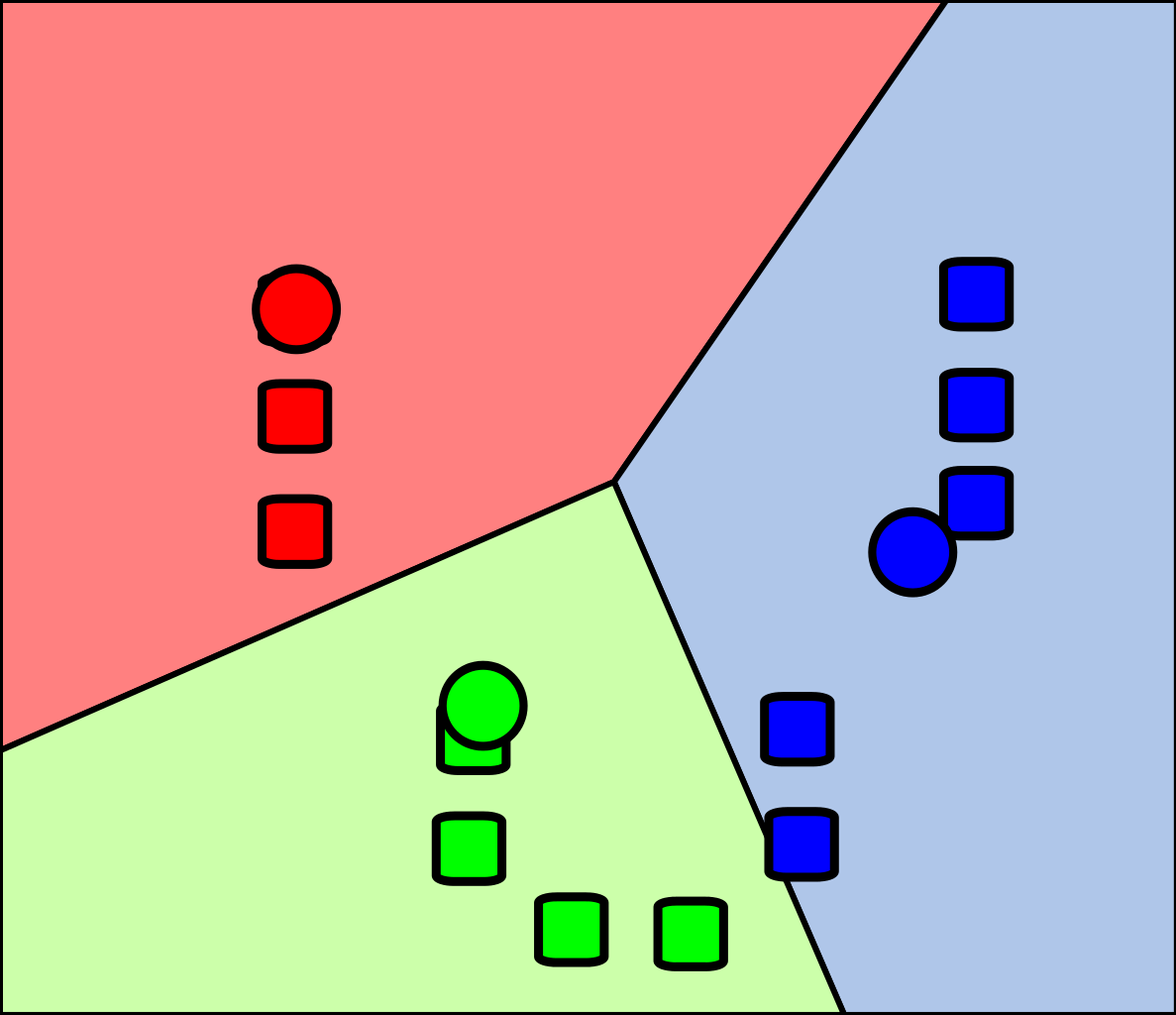
Comme illustré dans la figure suivante, la première étape du K-means consiste à placer dans l’espace des centroïdes au hasard. Ensuite, on associe chaque observation au centroïde le plus près de chacune :
Cette façon de fonctionner a l’avantage qu’elle garantit que les groupes formés minimisent la variance intra-groupe. Autrement dit, que les groupes formés soient le plus homogènes possible.
27.2.2 À propos du nombre de groupes
Vous avez peut-être remarqué dans l’exemple précédent que nulle part dans la procédure, l’algorithme du K-means change le nombre de groupes. C’est parce que, un peu comme pour le NMDS (Chapitre 26), nous devons spécifier au démarrage combien de groupes nous cherchons (qui se nomme ici k). L’algorithme fournira une solution différente selon le k choisit par l’utilisateur.
27.2.3 Le critère de Calinski
Donc, comment doit-on procéder si on ne sait pas à l’avance combien de groupes contient notre jeu de données? Comment peut-on savoir le plus objectivement possible combien de groupes il contient? L’outil pour y arriver se nomme le critère de Calinski.
Le critère de Calinski, un peu comme un ratio de F, se calcule en divisant la variance inter-groupe par la variance intra-groupe.
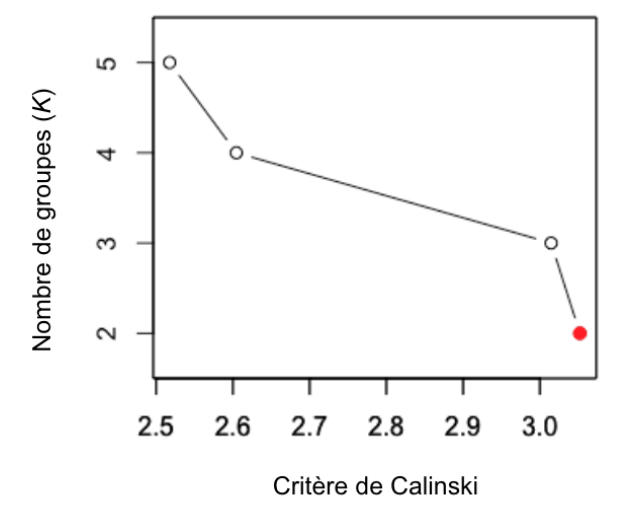
La stratégie pour déterminer le k optimal consistera donc à calculer un partitionnement avec différents k (p. ex. de k=2, k=3, etc. jusqu’à un maximum arbitraire, par exemple k=10) et d’observer l’évolution du critère de Calinski :
Le meilleur k sera celui avec la valeur de Calinski la plus élevée. Dans la figure précédente, le nombre de groupes optimal serait de 2.
Notez que ces groupes ne sont pas nécessairement les plus faciles à interpréter biologiquement ou écologiquement. Vous pouvez (fortement recommandé!) utiliser votre jugement à cette étape, particulièrement si les valeurs de Calinski sont très rapprochées entre certaines valeurs de k.
Notez aussi que cet indice est une façon parmi d’autres pour déterminer le k optimal. Il en existe aussi d’autres si vous fouillez un peu. Le critère de Calinski est le plus utilisé. Il est néanmoins peu fiable lorsque la taille des groupes est très inégale. Il faut dans ces cas se fier à notre jugement plutôt que de se fier aveuglément au critère.
27.2.4 La part du hasard
Comme pour la NMDS, l’algorithme du K-means démarre avec une configuration aléatoire. C’est donc dire que si vous relancez l’algorithme une seconde fois, il pourrait ne pas nécessairement trouver exactement le même résultat.
Il est généralement recommandé de lancer l’algorithme avec au moins 20 configurations de départ différentes (idéalement 50) et de conserver le meilleur résultat (i.e. la configuration avec le critère de Calinski le plus élevé). Ne vous en faites pas, la fonction R fera ces 50 répétitions automatiquement pour vous.
27.2.5 Labo : K-means en choisissant le k avant de débuter
Nous allons maintenant essayer d’appliquer la technique des K-means au jeu de données sur les manchots de Palmer, afin d’évaluer si l’algorithme réussit à séparer les espèces en se basant uniquement sur les mesures morphologiques.
Comme pour l’ACP, nous préparerons deux tableaux, soit un avec les variables quantitatives, et un autre avec les informations complémentaires que nous pourrons utiliser pour valider la classification.
library(tidyverse)
── Attaching core tidyverse packages ──────────────────
✔ dplyr 1.1.4 ✔ readr 2.1.5
✔ forcats 1.0.0 ✔ stringr 1.5.1
✔ ggplot2 4.0.1 ✔ tibble 3.3.0
✔ lubridate 1.9.4 ✔ tidyr 1.3.1
✔ purrr 1.0.4
── Conflicts ───────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()
ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errors
library(palmerpenguins)
Attaching package: 'palmerpenguins'
The following objects are masked from 'package:datasets':
penguins, penguins_raw
Remarquez que dans une vraie application, il aurait été prudent d’explorer nos données avant de commencer, mais comme nous connaissons bien le jeu de données penguins, inutile de répéter ce travail ici.
Ensuite, ajustons un premier modèle de K-means, avec k=3 pour voir à quoi les résultats d’un K-means peuvent ressembler :
library(vegan)
Loading required package: permute
Loading required package: lattice
exemple <- pour_regroupements |>scale() |>kmeans(centers =3, nstart =50)
Il y a plusieurs choses importantes dans ce bout de code. D’abord, il faut centrer-réduire chacune de nos variables avant le calcul (la fonction scale) afin d’éviter les problèmes d’échelles, puisque le k-means travaille avec la distance euclidienne. Aussi, la fonction à utiliser pour faire le calcul se nomme kmeans. Elle attend 3 arguments, soit le tableau de données (implicite dans la chaîne), le nombre de groupes (k) et le nombre de configuration de départ à essayer (nstart).
Voyons maintenant ce que contient notre objet de résultats :
On apprend d’abord dans ces sorties que la technique a produit 3 groupes (comme on avait demandé), qui contiennent 123, 87 et 132 observations chacun.
La section Cluster means nous informe de la moyenne de chacune des variables pour chacun des groupes. On voit par exemple que les manchots du premier groupe ont des becs plus longs (0,65) et moins épais (-1,09) que la moyenne.
Rappelez-vous qu’on ne parle plus ici de mm, puisque les données ont été centrées-réduites. On parle plutôt de 0,65 écart-type au-dessus de la moyenne.
La section Clustering vector nous informe ensuite du groupe auquel appartiennent chacune des observations après le classement.
Les dernières lignes nous informent enfin de certaines statistiques sur le partitionnement de la variance que nous n’utilisons pas ici.
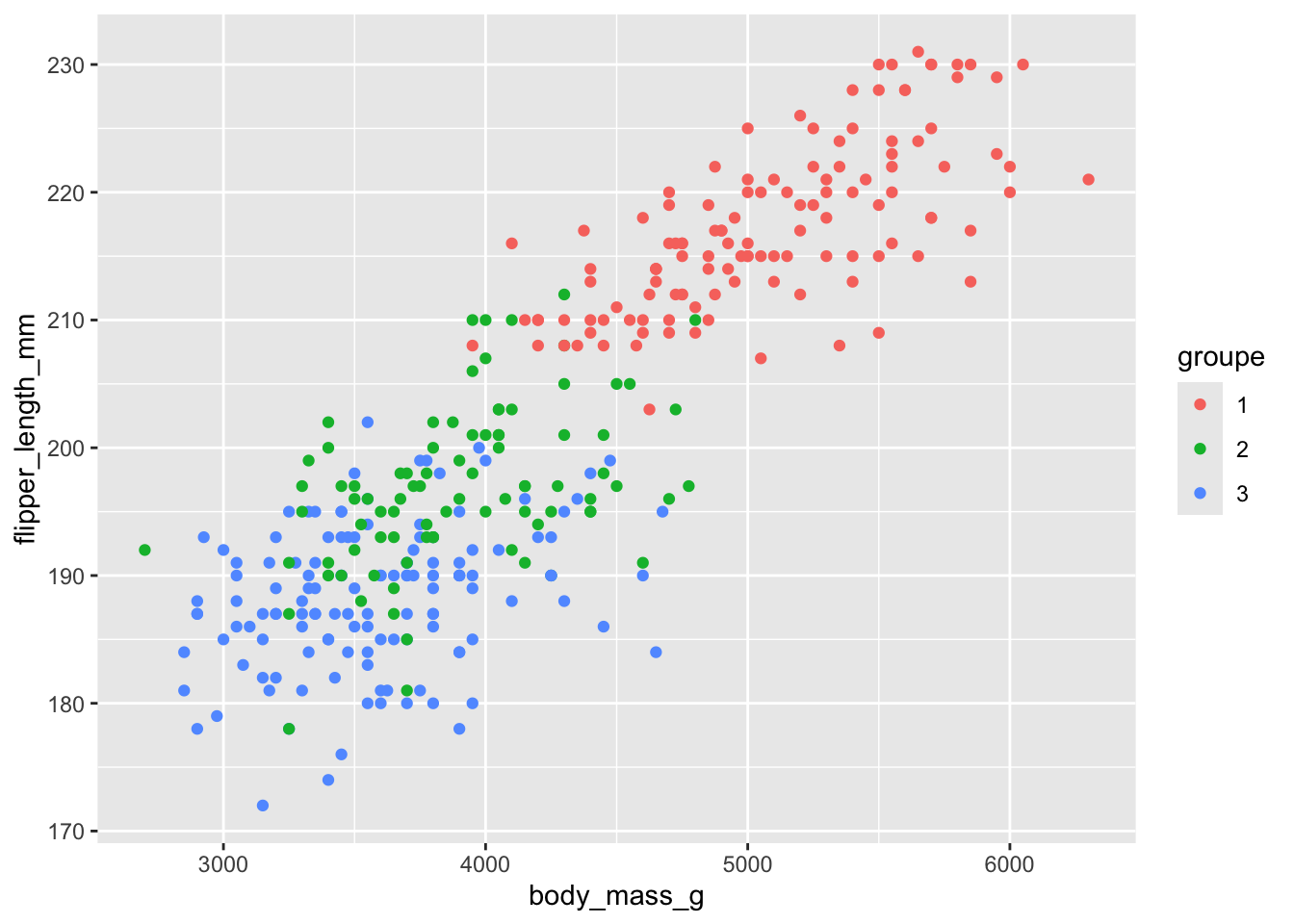
Voyons maintenant comment on pourrait se faire un petit graphique pour voir comment se répartissent nos groupes. La clé pour y arriver est d’ajouter à notre tableau de données une colonne de groupe, que l’on extrait de notre objet de résultats. On peut ensuite utiliser cette information de groupe pour colorer les éléments dans notre graphique.
Remarquez que l’on doit utiliser la fonction as_factor pour emballer la colonne de groupe, car sinon, R l’aurait interprétée comme une variable quantitative puisque ce sont des chiffres.
Remarquez aussi que sur ces 2 variables, on voit bien la différence entre le groupe 1 et les groupes 2 et 3, mais que ces derniers sont très difficiles à distinguer.
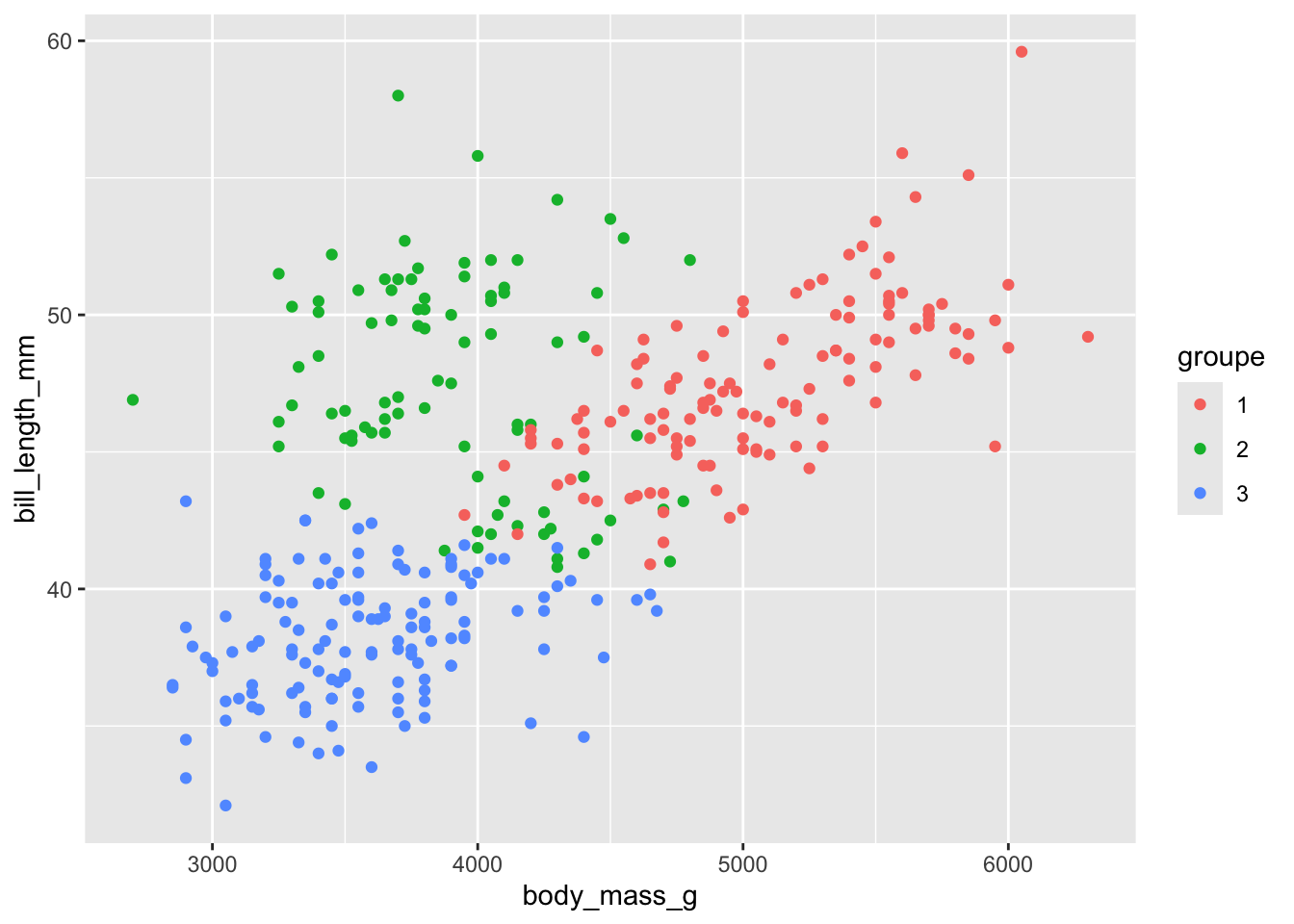
En se fiant sur les sorties numériques, on pourrait par exemple aller explorer la longueur du bec / poids du corps pour mieux illustrer les 3 groupes :
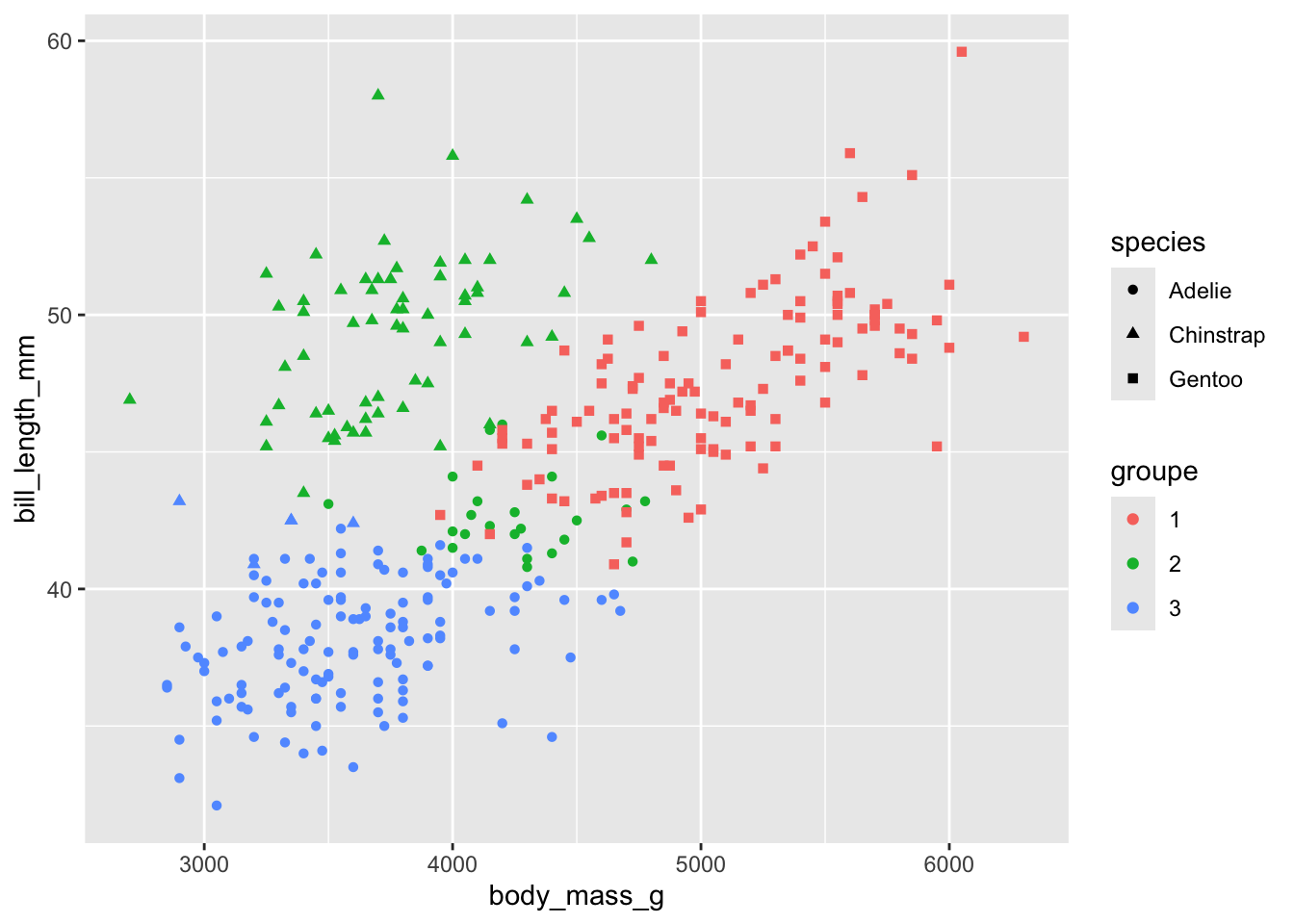
Enfin, on pourrait aller voir à quel point le k-means a réussi à retrouver la séparation originale entre les 3 espèces, pour laquelle il n’avait pas l’information pour se valider.
On voit que tous les Gentoo se sont retrouvés dans le groupe 1. La majorité des Adélie dans le groupe 3 et la majorité des manchots Chinstrap dans le groupe 2.
Évidemment ce n’est pas parfait, mais l’algorithme des k-means n’est pas fait pour reclasser parfaitement les groupes. Il est fait pour trouver les groupes les plus naturels dans nos données…
27.2.6 Labo : K-means pour sélectionner le meilleur nombre de groupes
Comme expliqué plus haut, dans la vraie vie, on ne saura pas toujours combien de groupes contiennent nos données. Par exemple, si on applique un k-means sur les données physicochimiques d’une série de lacs, on ne saura pas d’avance en combien de groupes nos lacs devraient être séparés.
Pour déterminer le nombre de groupes idéal dans un jeu de données, la fonction pour y arriver provient aussi de la librairie vegan et se nomme cascadeKM. Elle fonctionne un peu comme kmeans, mais on lui fournit un k minimum et un k maximum, et elle essaie elle-même tous les k possibles dans cet intervalle. Si on voulait par exemple essayer entre 2 et 5 groupes, on l’utiliserait comme ceci :
plusieurs <- pour_regroupements |>scale() |>cascadeKM(inf.gr =2, sup.gr =5, iter =50)
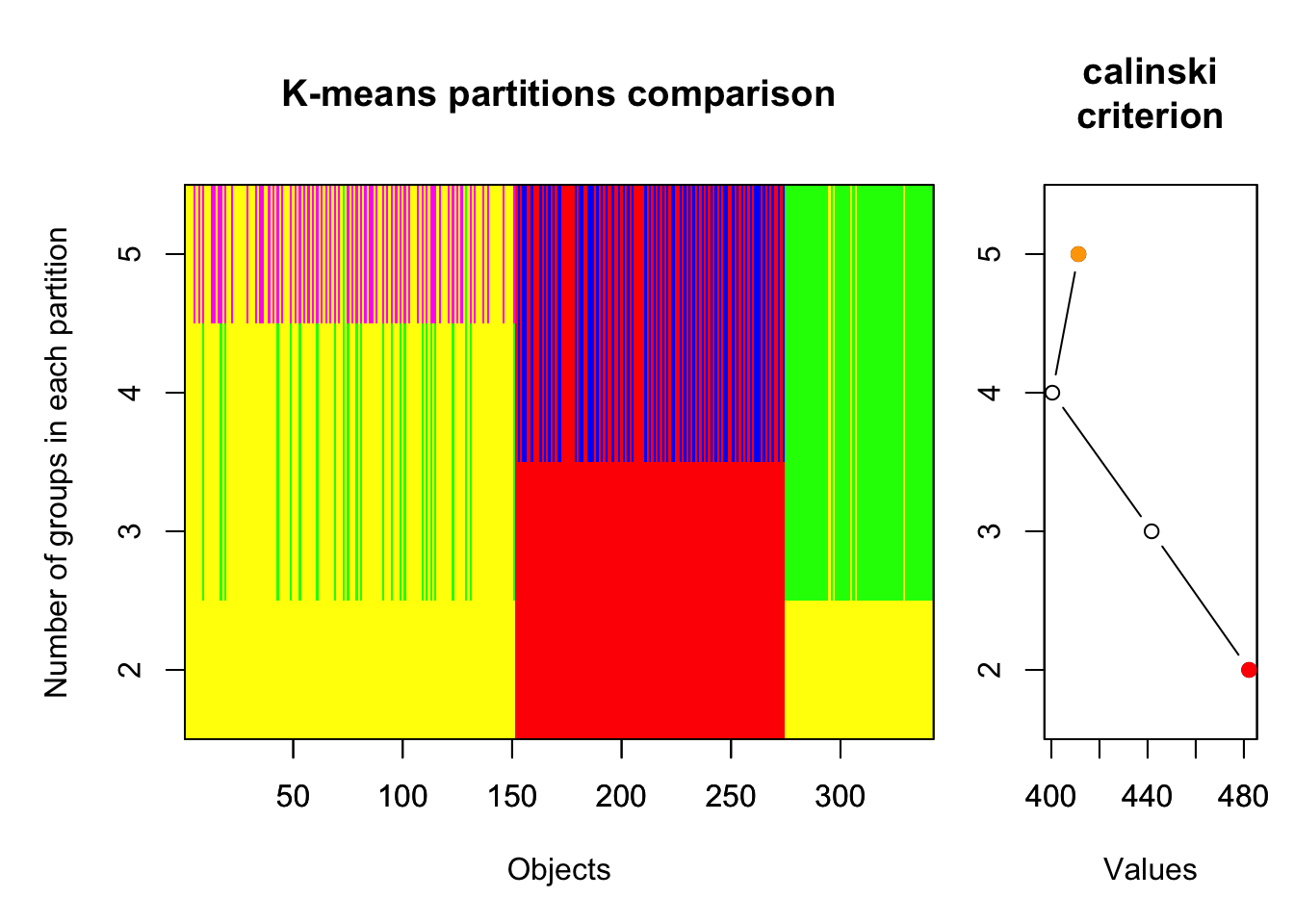
Par la suite, on peut explorer les valeurs du critère de Calinski pour ces résultats à l’aide de la fonction plot :
plot(plusieurs)
La partie de gauche ferait une excellente peinture abstraite (!), mais elle nous montre surtout à quel groupe (la couleur) appartient chacune des observations (en X) selon la valeur de k (en Y). La partie de droite nous montre quant à elle le critère de Calinski calculé (en X) pour chacune des valeurs de k (en Y). On y voit que selon le critère de Calinski, le nombre de groupes idéal serait de 2, puisque c’est la valeur la plus élevée.
On peut aussi accéder à ces résultats en chiffres, en tapant le nom de notre objet de résultats dans la console :
plusieurs$results
2 groups 3 groups 4 groups 5 groups
SSE 564.0535 378.2832 299.5212 231.9172
calinski 482.1915 441.6771 400.4100 411.2587
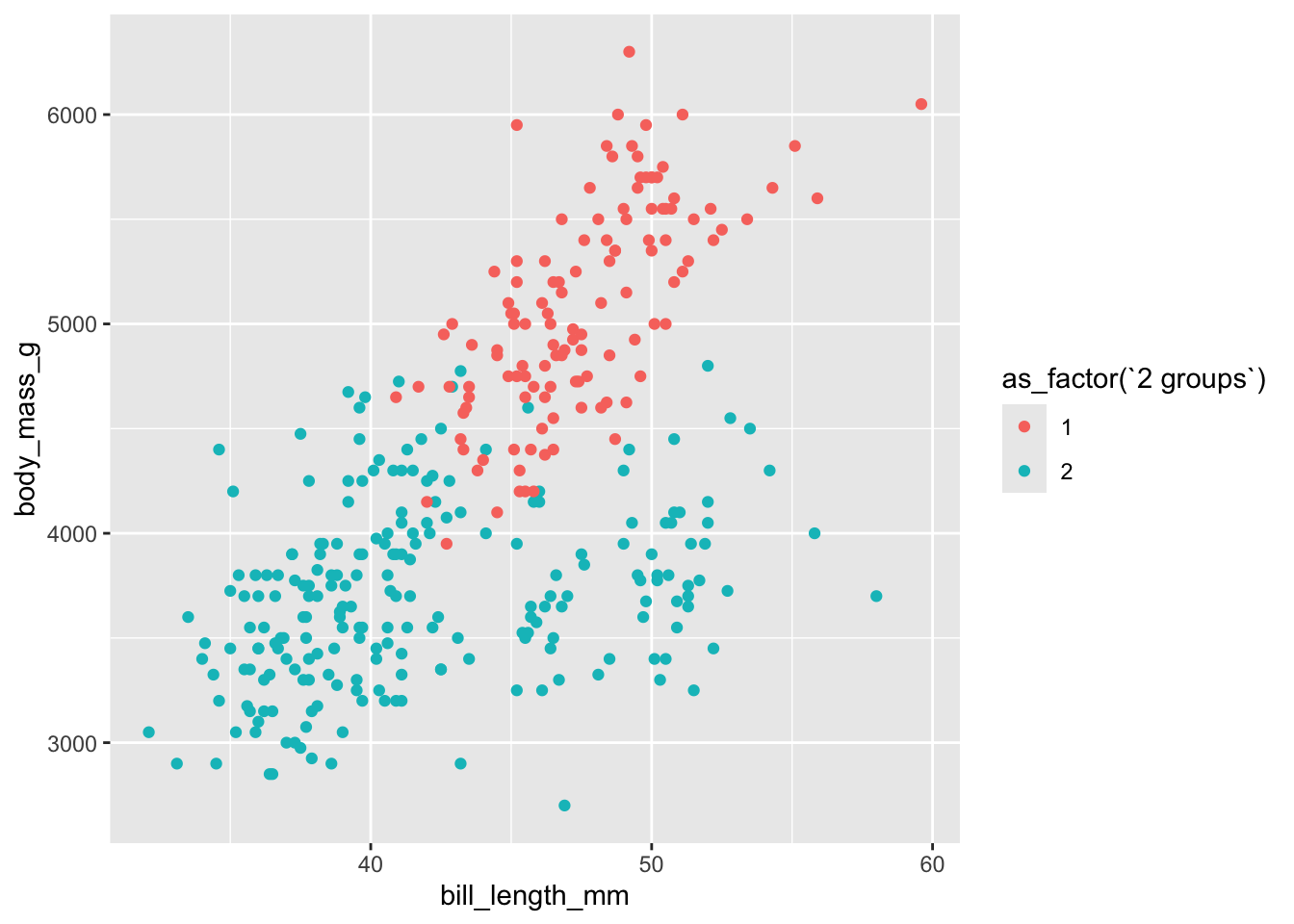
Si on voulait explorer visuellement à quoi ressemble la classification à 2 groupes, on pourrait ajouter à notre tableau de données le résultat de la classification, et l’utiliser dans notre graphique pour choisir la couleur comme dans le graphique précédent. Il faudrait par contre ici utiliser la fonction bind_cols, puisque l’on connectera plusieurs colonnes de résultats à la fois.
Remarquez dans le code précédent que nous avons dû utiliser les backticks (`) pour accéder au nom de la colonne désirée, soit celle pour 2 groupes, puisque son nom contient des espaces.
27.3 La classification hiérarchique
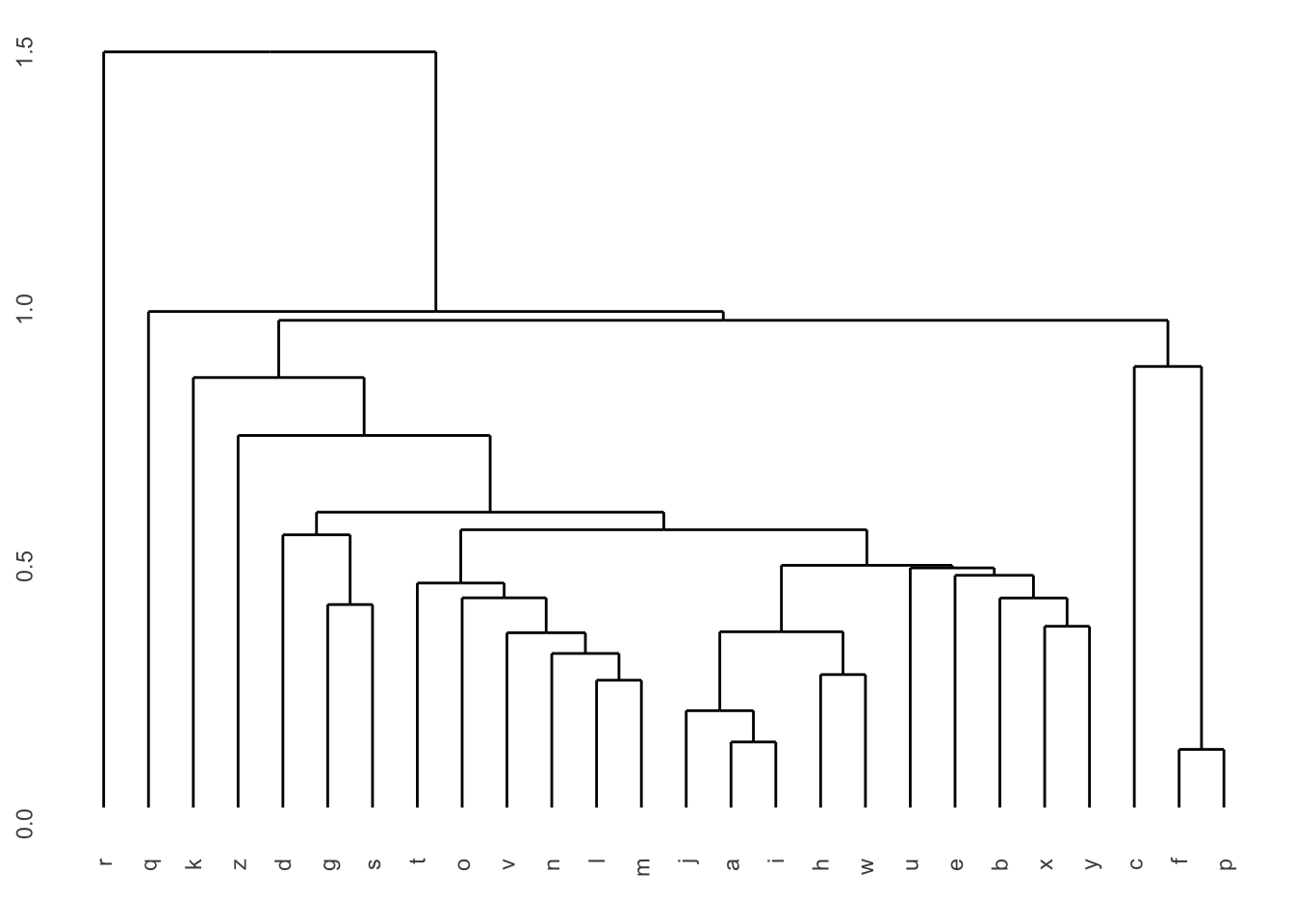
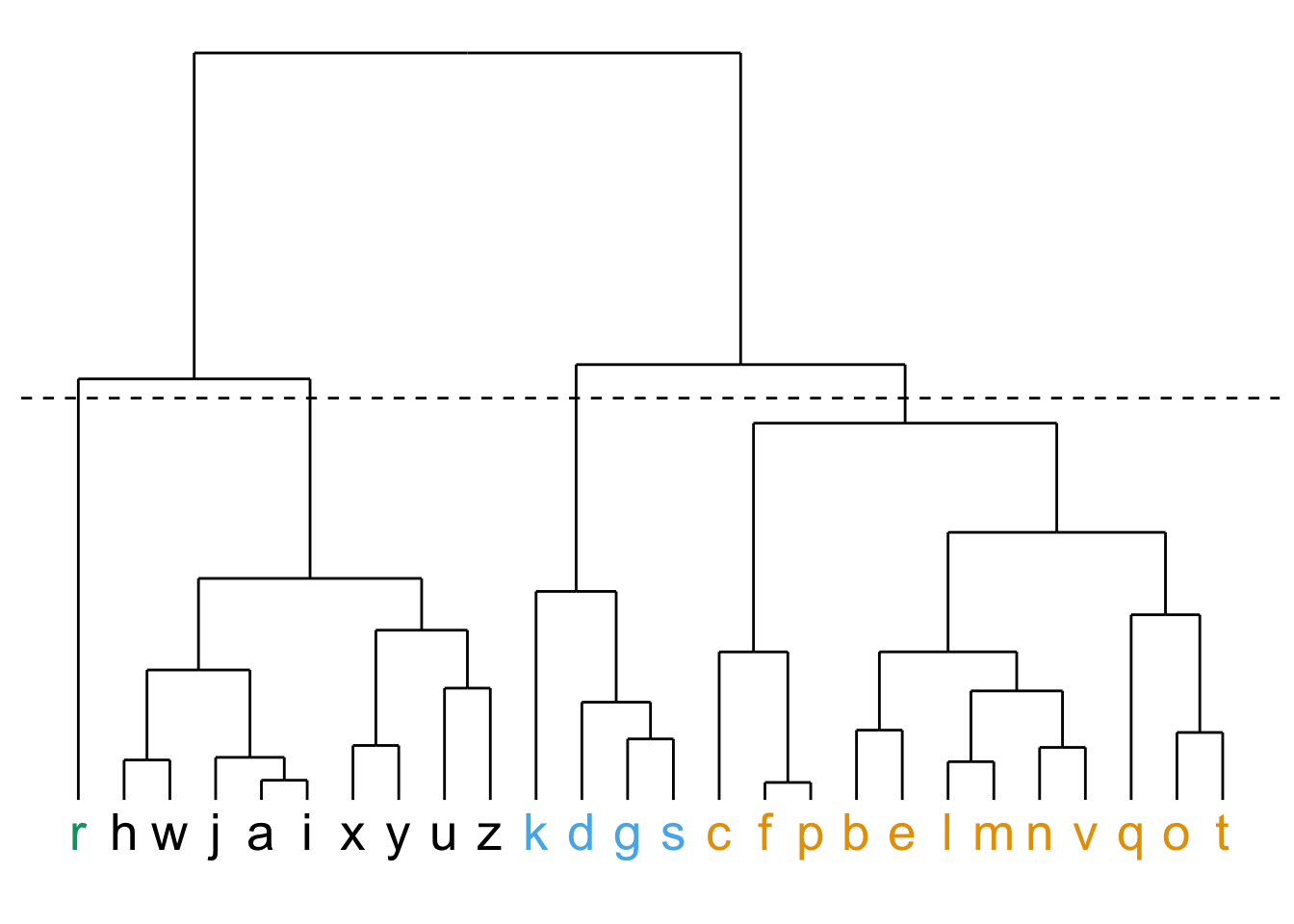
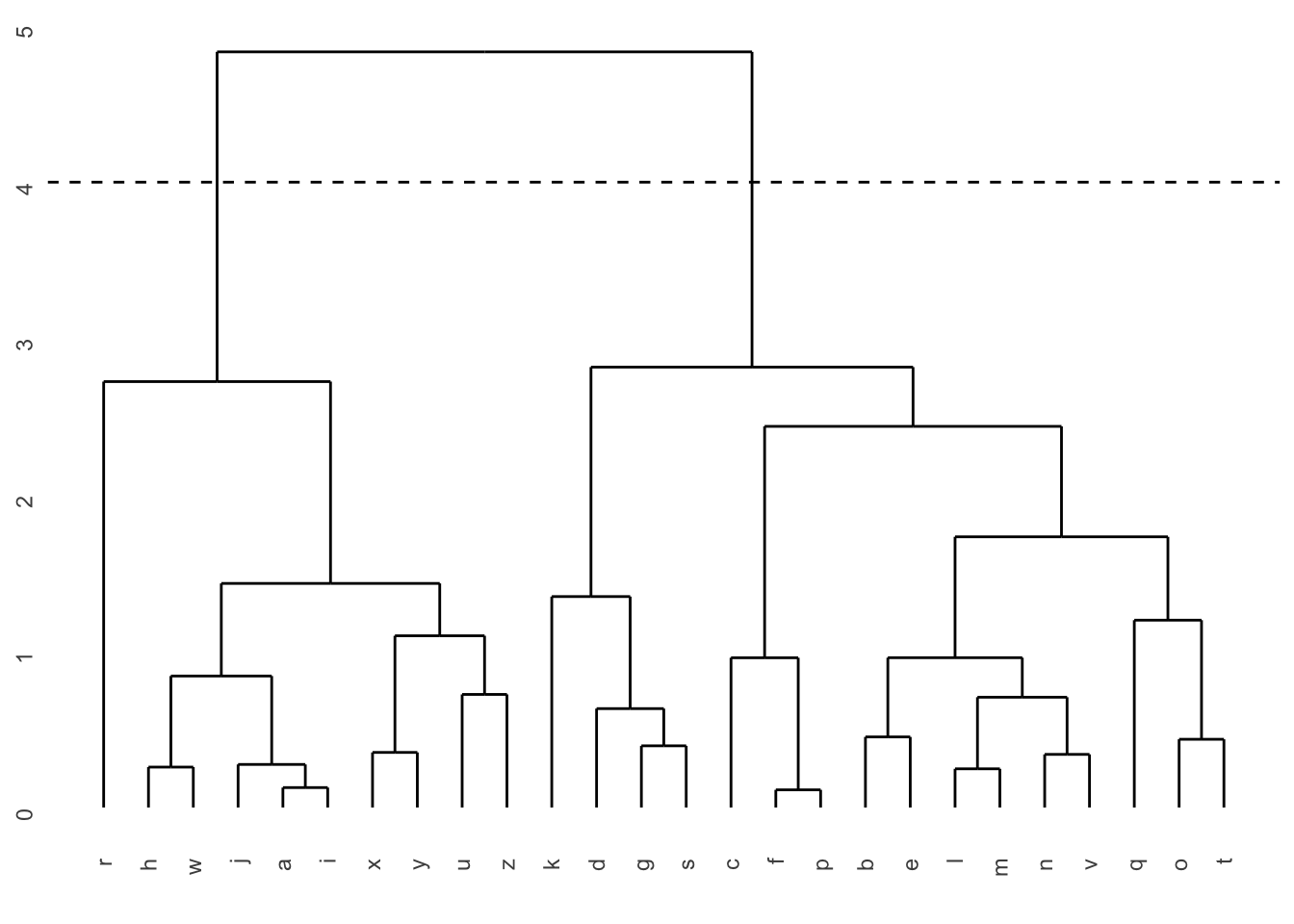
Pour comprendre la classification hiérarchique, observons-en d’abord la sortie, que l’on nomme un dendrogramme :
Dans cette figure, l’axe des X représente chacune des observations (dans un ordre quelconque; dans un cas réel, nous aurions affiché le nom de chaque observation au bout de la ligne… ). L’axe des Y est un axe de distance (p. ex. euclidienne) entre les observations ou les groupes.
Plus la connexion entre deux observations est haute sur l’axe des Y, plus ces observations (ou ces groupes) sont différentes les unes des autres. On obtient donc une vue d’ensemble des ressemblances entre nos observations.
Notez que ultimement, toutes les observations sont connectées.
27.3.1 Fonctionnement de l’algorithme
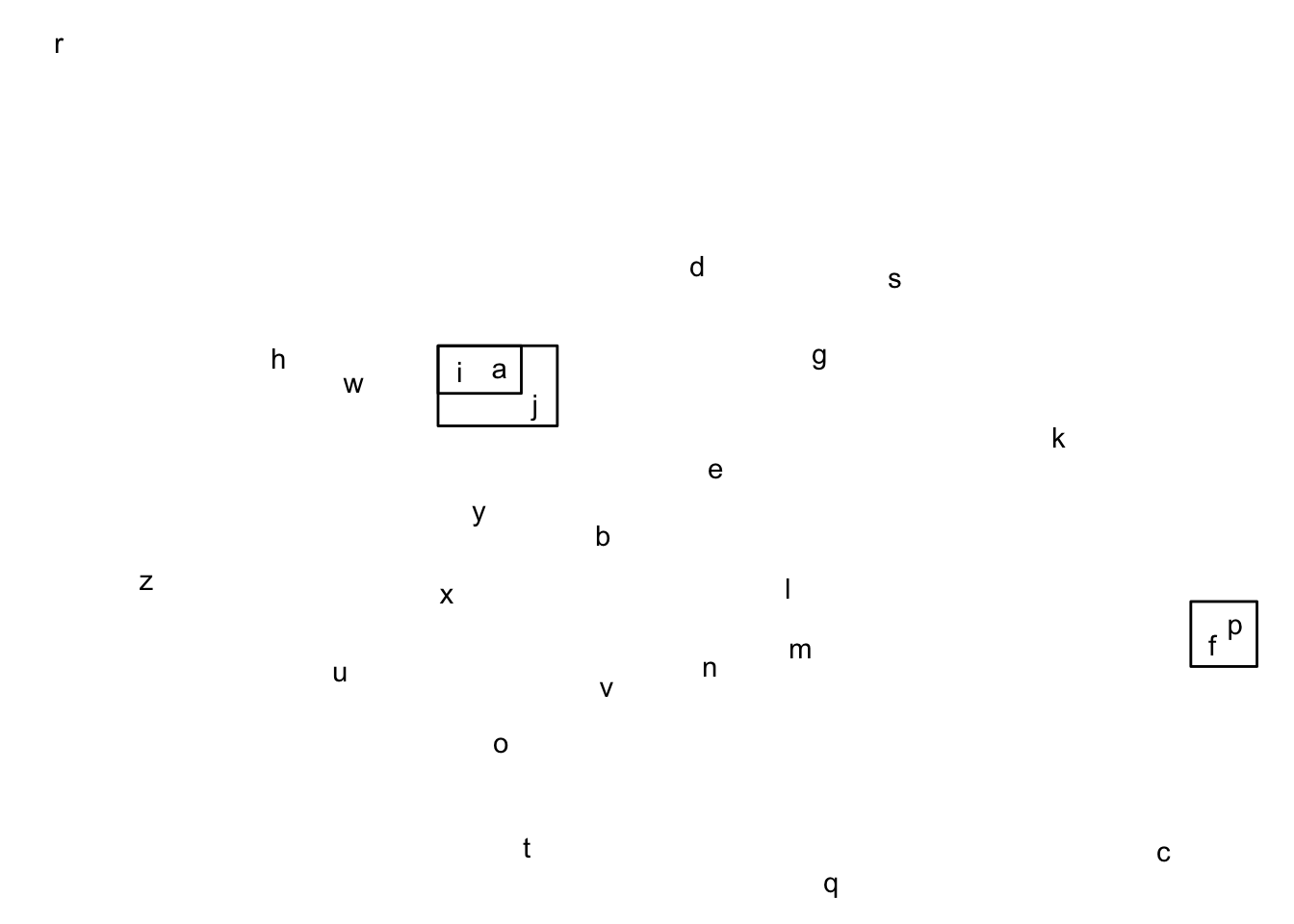
Au début de la procédure de partitionnement hiérarchique, l’algorithme considère chaque observation comme un groupe, contenant une seule observation.
Avant de débuter
Connecter p et f
Connecter i et a
Connecter j à i+a
L’algorithme va tout d’abord calculer une matrice de distances entre ces groupes (exactement comme au Chapitre 22).
Il va ensuite trouver la paire de groupes la plus proche et les connecter.
Il recalcule ensuite une nouvelle matrice de distances entre les groupes, puis reconnecte les deux groupes les plus proches.
Il continue ainsi jusqu’au moment où toutes les observations sont connectées.
Autrement dit, le dendrogramme est construit par le bas, et on remonte vers le haut.
27.3.2 Combien de groupes
Comme pour le K-means, on peut ensuite se demander : oui mais, combien de groupes contient notre jeu de données finalement?
Encore une fois, il n’y a pas de réponse absolue à cette question.
La façon habituelle de faire est de choisir un seuil de distance à partir duquel on considère les groupes comme différents. Dans la partie de gauche de la figure précédente, en établissant notre seuil de distance à 4, on obtient 2 groupes, alors qu’en le plaçant à 2,6, on en obtient 4.
Plus on choisit un seuil de distance élevé, moins notre solution contiendra de groupes.
Il existe certaines règles du pouce (semblables au critère de Calinski) pour choisir le nombre final de groupes, mais cette tâche est souvent effectuée à l’œil, de façon arbitraire.
27.3.3 Le choix de la mesure d’attachement
Un point sur lequel nous avons glissé au moment de définir l’algorithme est de savoir comment on mesure la distance entre deux groupes. Il faut par contre bien faire la nuance entre deux concepts.
Le terme distance, dans ce contexte, est consacré à la mesure de distance, comme décrit au Chapitre 22 (euclidienne, Bray-Curtis, etc.)
Le terme attachement (linkage) définit par quels points on mesure la distance entre deux groupes. Est-ce qu’on calcule la distance par le point le plus loin du groupe, le plus proche, le milieu du paquet, etc?
Comme d’habitude, il n’existe pas de façon magique, meilleure que toutes les autres pour faire cette tâche. Il en existe des dizaines, mais nous en verrons 3 :
La méthode complète (complete) se base sur la distance entre les deux points les plus éloignés
La méthode simple (single) se base sur la distance entre les deux points les plus proches
La méthode moyenne (average) se base sur la distance moyenne entre toutes les paires de points
En général, la méthode simple aura tendance à attacher les observations une à une, formant des groupes qui ne sont pas très compacts.
La méthode simple (single)
La méthode complète créera des groupes compacts, mais peu séparés les uns des autres.
La méthode complète (complete)
La méthode moyenne présente en général un bon compromis, formant des groupes à la fois relativement compacts et espacés.
La méthode moyenne (average)
Par contre, les méthodes simple et complète offrent une interprétation simple du point de coupure, que la méthode moyenne n’offre pas. Si l’on place par exemple notre point de coupure à une distance euclidienne de 4, comme ceci :
Dans la méthode simple, on peut affirmer que chaque point dans un groupe possède au moins un autre point à une distance de moins de 4.
Dans la méthode complète, on peut affirmer que tous les points d’un groupe sont à moins de 4 les uns des autres.
Dans la méthode moyenne, il n’y a pas d’interprétation de ce genre possible.
Aussi, contrairement aux deux autres méthodes, la méthode moyenne est sensible aux transformations. Si vous transformez vos données, elle pourrait vous donner un résultat différent.
27.3.4 Le choix de la mesure de distance
Contrairement au K-means qui fonctionne obligatoirement avec la distance euclidienne, la classification hiérarchique nous permet de choisir la mesure de distance désirée. Tout ce que nous avons vu au Chapitre 22 s’applique donc ici :
si vos données sont continues, on utilise la distance euclidienne,
si vos données sont des décomptes d’individus, on utilise la distance de Bray-Curtis et
si vous avez des données de présence-absence, vous pouvez utiliser la distance de Jaccard ou celle de Bray-Curtis.
Il est aussi important de s’interroger sur les différences de variance et de standardiser les données si nécessaire.
27.3.5 Labo : La classification hiérarchique
Comme nous avons vu plus haut, le dendrogramme du partitionnement hiérarchique affiche chacune des obsevations au bas du graphique. Pour que notre graphique demeure lisible dans les pages de ce livre, nous allons piger un petit échantillon aléatoire dans le tableau de données des manchots et travailler uniquement avec ce dernier. Il faut aussi penser de réduire notre tableau d’informations supplémentaires pour qu’il contienne lui aussi uniquement les lignes choisies.
La fonction pour construire un dendrogramme de classification hiérarchique se nomme hclust (Hiearchical CLUSTering), et provient aussi de la librairie vegan. Par contre, cette fonction ne s’attend pas à recevoir un tableau de données brutes, mais plutôt une matrice de distances entre les observations. C’est pourquoi nous allons construire une chaîne, dans laquelle nous allons aussi utiliser la fonction vegdist, pour calculer une matrice de distances. Comme nous utiliserons la distance euclidienne, il faut aussi centrer et réduire nos données avant de procéder au calcul :
On peut ensuite visualiser notre dendrogramme à l’aide de la fonction ggdendrogram de la librairie ggdendro :
library(ggdendro)ggdendrogram(dendro)
Il existe aussi une fonction de base (plot) pour afficher le dendrogramme, mais cette dernière n’est pas basée sur ggplot2.
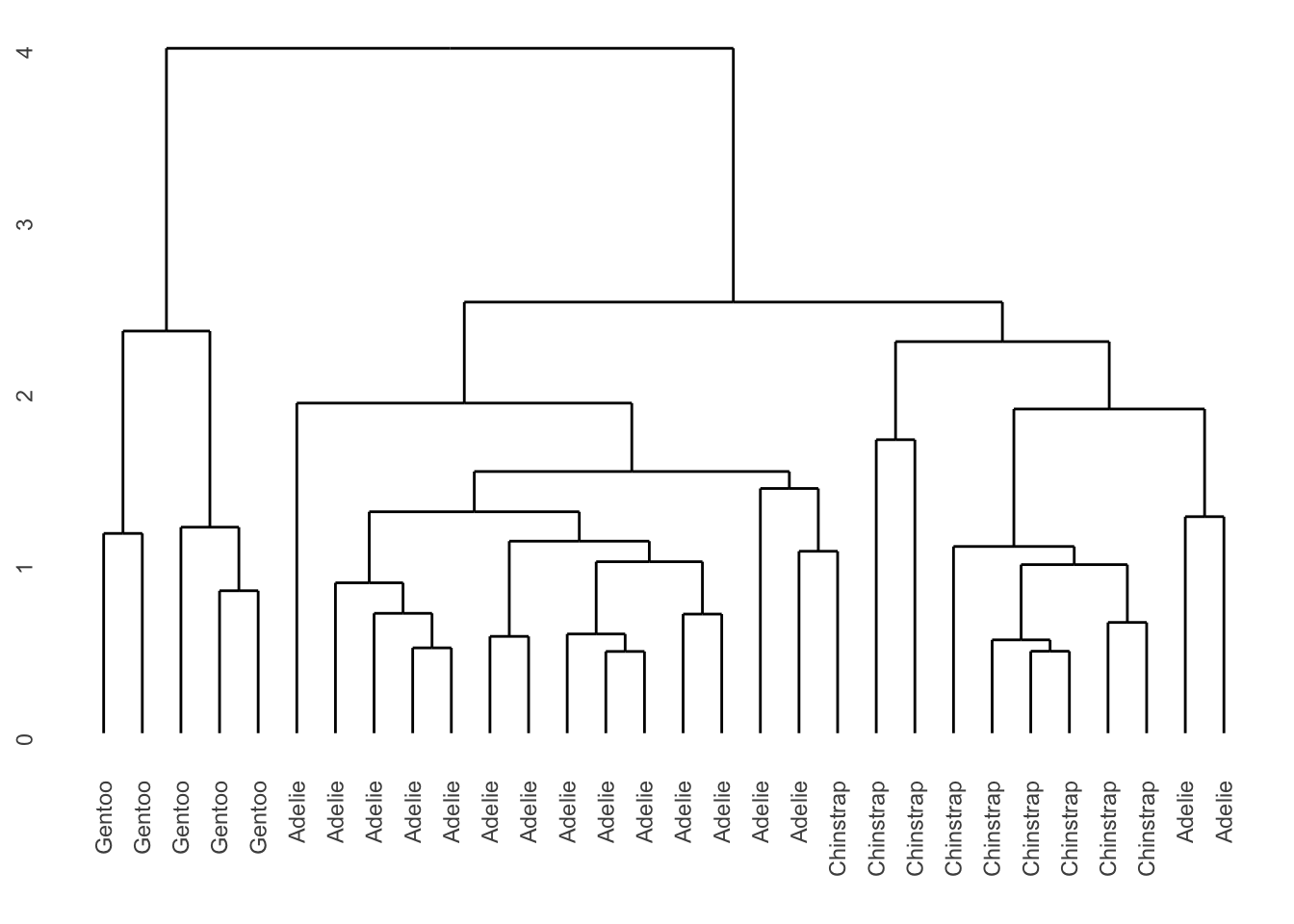
On pourrait aussi inscrire au bas du graphique une information plus intéressante que le numéro de la ligne de chaque observation, par exemple, l’espèce de chaque individu.
Pour se faire, il faut ajouter manuellement l’étiquette à chacune des lignes de notre dendrogramme. Comme l’objet dendro n’est pas un tableau de données, on ne peut PAS utiliser la fonction mutate :
dendro$labels = infos_pour_hclust$species
On peut ensuite afficher le dendrogramme :
ggdendrogram(dendro)
La première séparation, une distance d’environ 4, sépare 5 manchots Gentoo du reste du groupe. La deuxième séparation, à une hauteur d’environ 2,5 sépare les manchots Adélie des manchots Chinstrap et d’un groupe de deux manchots Adélie un peu plus différents à droite.
Comme discuté ci-haut, le partionnement hiérarchique ne fournit pas de nombre optimal de groupes. Ce serait à nous, à l’oeil, de déterminer le meilleur nombre de groupes.
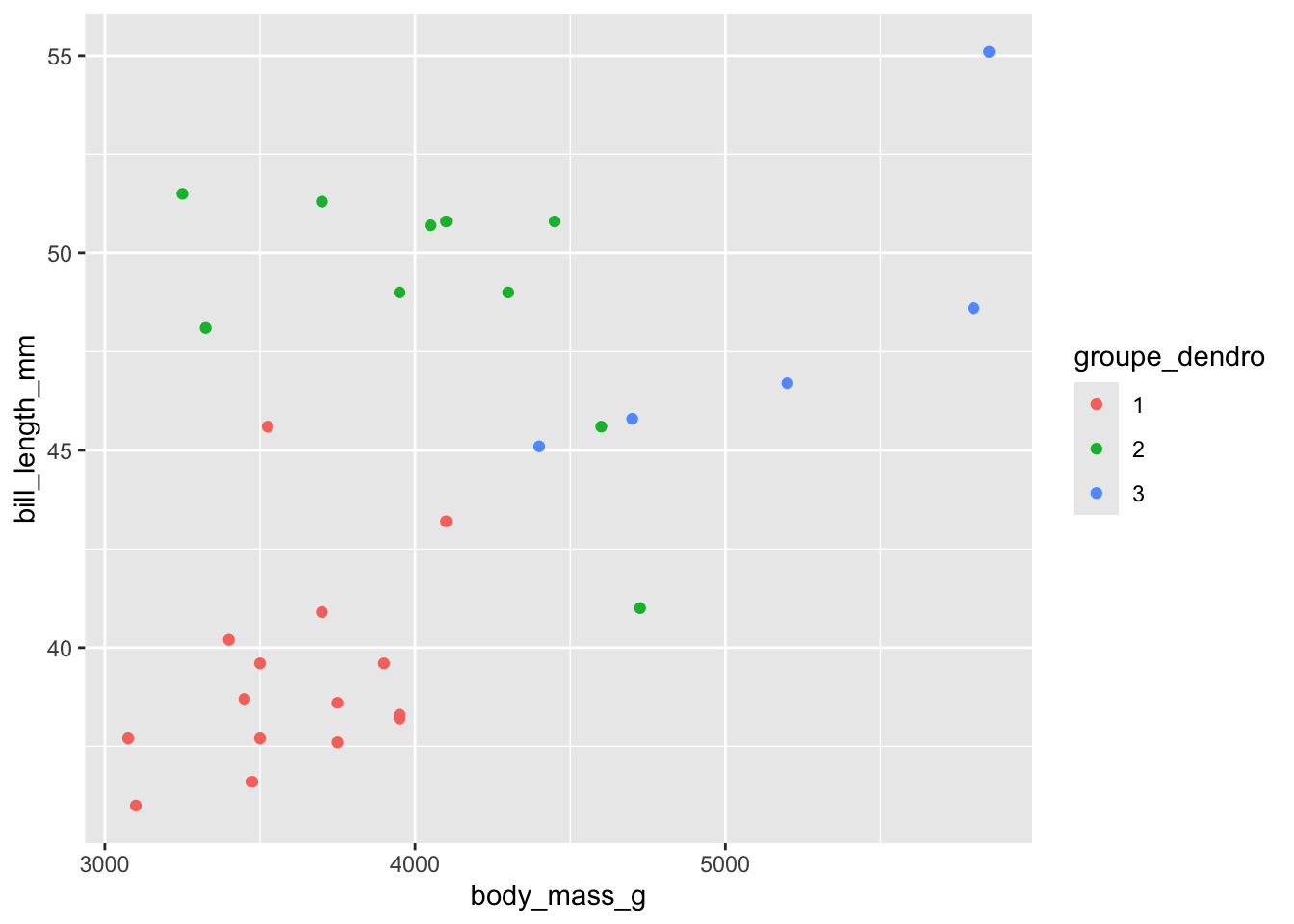
Si on voulait explorer la classification donnée par 3 groupes dans ce dendrogramme, on peut utiliser la fonction cutree, et lui fournir le nombre de groupes désiré. La fonction va elle-même aller couper l’arbre et nous dire dans quel groupe tomberait chacune de nos observations.
trois <-cutree(dendro, k=3)pour_hclust |>mutate(groupe_dendro =as_factor(trois)) |>ggplot(aes(body_mass_g, bill_length_mm)) +geom_point(aes(color = groupe_dendro))
27.4 Exercice : Les analyses de regroupements
À partir de la base de données de météo des villes utilisée au Chapitre 231, nous allons maintenant appliquer les techniques ci-haut pour tenter de voir si on trouve des regroupements naturels entre les villes, basé sur la météo.
Comme au Chapitre 23, éliminez la variable de neige avant de commencer les analyses puisque celle-ci contient beaucoup de valeurs manquantes.
Appliquez dans un premier temps la technique des K-means pour déterminer le meilleur nombre de groupes que pourrait contenir ce jeu de données en vous basant sur le critère de Calinski.
Ensuite, illustrez ce partitionnement à l’aide d’un graphique où on retrouvera en X la température maximum moyenne et en Y les précipitations en mm.
Sur quel critère semble s’être basé le K-means pour séparer nos groupes? Géographiquement, qu’ont en commun les villes dans le groupe de gauche du graphique?
Ensuite, appliquez la technique du partitionnement hiérarchique au même jeu de données et produisez le graphique du dendrogramme.
Quelles sont les villes appartenant au petit groupe se distinguant le plus du reste des villes?
En général, appréciez-vous les groupes formés par le dendrogramme? Vous semblent- ils naturels?
27.5 Conclusion
La question qui vous reste en tête à ce moment-ci est probablement : OK, parfait Charles, on a deux techniques pour explorer les regroupements, mais laquelle on utilise dans quelle situation?
En général, la technique des K-means sera utilisée lorsque vous connaissez à l’avance le nombre de groupes, mais que vous ne connaissez pas les règles exactes qui permettraient d’automatiser le processus de classification. Cette technique est très utilisée par exemple pour séparer les différents types de terrain dans les applications de télédétection. Le K-means est plutôt une technique d’apprentissage automatique (machine learning) où on laisse l’ordinateur démêler les données pour nous.
Le partitionnement hiérarchique quant à lui est surtout utilisé dans des applications d’écologie des communautés, où on tente de comprendre quels sites se ressemblent par leur composition en espèces. C’est une technique plus exploratoire, mais qui permet aussi de meilleures interprétations biologiques des sorties que le K-means, particulièrement grâce à l’aspect hiérarchique des sorties.
Ici, pour des raisons pédagogiques, je vous ai montré les deux techniques sur un même problème, mais dans la vraie vie, on en choisit une ou l’autre, selon le travail à faire.





{kind=link}
{kind=link}
{kind=link}
{kind=link}